What Is a Macular Hole and How Does It Develop?
18/05/2026

06/05/2026
Inherited retinal disease (IRD) encompasses a diverse group of disorders characterised by dysfunction and degeneration of the retina due to genetic mutations. Although individually rare, collectively they represent a significant cause of visual impairment worldwide, particularly among younger patients. These conditions are often described under the broader umbrella of retinal dystrophy, reflecting the progressive and degenerative nature of the pathology.
For many patients, the journey begins subtly. Early symptoms may include difficulty seeing in dim light, commonly referred to as night blindness, or mild visual disturbances that are easy to overlook. Over time, however, these symptoms can evolve into more pronounced visual impairment, including constricted visual fields, reduced central vision, and ultimately progressive vision loss that significantly impacts daily life.
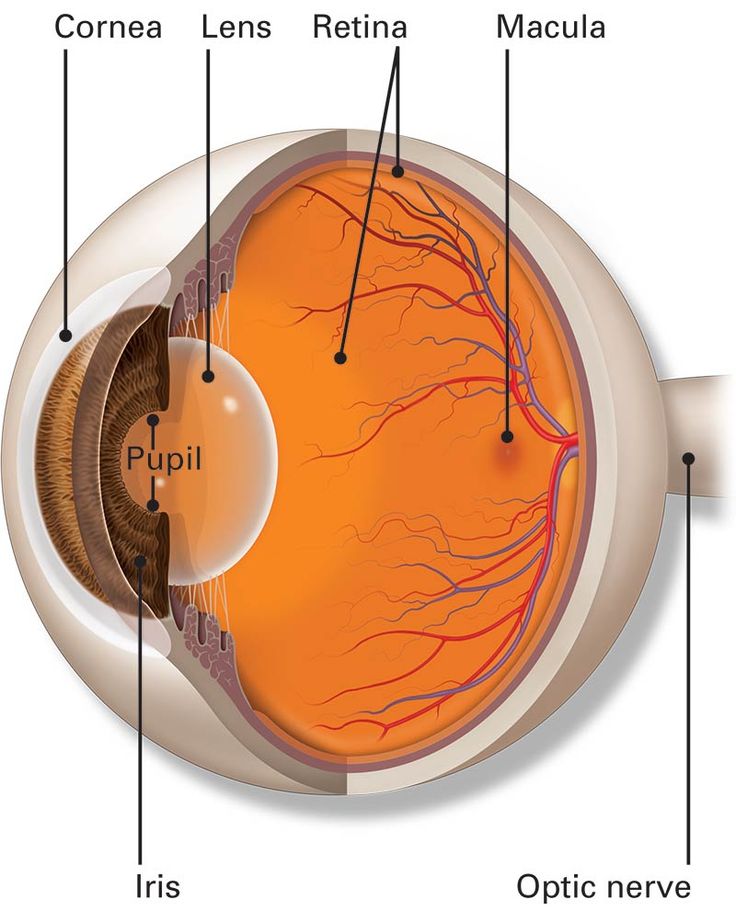
The retina is a highly specialised neural tissue, composed of photoreceptors (rods and cones), bipolar cells, ganglion cells, and supporting structures such as the retinal pigment epithelium. Each component plays a critical role in converting light into neural signals that are transmitted to the brain. In inherited retinal disease, mutations in genes responsible for these processes disrupt normal retinal function, leading to gradual cellular degeneration.
The genetic heterogeneity of IRD is remarkable. To date, hundreds of genes have been implicated, each associated with different clinical phenotypes. Some conditions primarily affect rod photoreceptors, leading to early night blindness and peripheral vision loss, as seen in retinitis pigmentosa. Others predominantly involve cone photoreceptors, resulting in early central vision impairment, colour vision defects, and photophobia, as observed in cone dystrophies and macular dystrophies. In many cases, both rods and cones are eventually affected, contributing to the characteristic pattern of progressive vision loss.
Clinically, IRD is often described as a family eye disease, reflecting its hereditary nature. However, the pattern of inheritance can vary widely, including autosomal dominant, autosomal recessive, X-linked, and mitochondrial transmission. This variability has important implications for diagnosis, prognosis, and genetic counselling. A detailed family history remains a cornerstone of clinical assessment, although the absence of a known family history does not exclude a genetic cause, particularly in recessive conditions.
Patients may present at different stages of disease, and the clinical findings can vary accordingly. In early stages, fundoscopic examination may appear relatively normal, despite significant functional impairment. As the disease progresses, characteristic changes may emerge, such as bone-spicule pigmentation, retinal vessel attenuation, and optic disc pallor in retinitis pigmentosa, or macular atrophy in certain macular dystrophies. Multimodal imaging, including optical coherence tomography and fundus autofluorescence, provides valuable insights into retinal structure and disease progression.
Electrophysiological testing, particularly electroretinography (ERG), remains an important diagnostic tool. It allows objective assessment of rod and cone function, helping to differentiate between various forms of retinal dystrophy. In many cases, ERG abnormalities precede visible structural changes, making it particularly useful in early disease.
Genetic testing has become an integral part of the diagnostic pathway for inherited retinal disease. By identifying the underlying mutation, clinicians can confirm the diagnosis, refine prognosis, and guide management. Moreover, genetic testing enables accurate counselling regarding inheritance patterns and recurrence risk within the family. For patients with a family eye disease, this information is often as important as the diagnosis itself.
The impact of inherited retinal disease extends beyond visual function. Patients frequently face significant psychological and social challenges, particularly as the disease progresses. The gradual nature of progressive vision loss can be difficult to cope with, affecting independence, employment, and quality of life. Early recognition and supportive care, including low vision rehabilitation and psychological support, are therefore essential components of management.
From a therapeutic standpoint, the landscape is evolving. Historically, treatment options for IRD were limited to supportive measures. Today, advances in molecular medicine are opening new possibilities. Gene therapy, in particular, has emerged as a promising approach for selected conditions. By delivering a functional copy of the defective gene, it is possible to restore or preserve retinal function in certain cases. Although currently available for only a limited number of genetic mutations, ongoing research is expanding the range of treatable conditions.
Other therapeutic strategies under investigation include gene editing, RNA-based therapies, and cell-based approaches such as retinal pigment epithelium transplantation. While many of these remain in experimental stages, they represent a shift towards targeted, disease-modifying treatments that were unimaginable just a decade ago.
In parallel, advances in imaging and functional testing are improving our ability to monitor disease progression and assess treatment response. Quantitative OCT metrics, adaptive optics imaging, and microperimetry provide increasingly detailed insights into retinal structure and function. These tools are not only valuable in clinical practice but also essential for the design and evaluation of clinical trials.
In specialised centres such as Barraquer Eye Hospital, the management of inherited retinal disease is approached in a multidisciplinary manner. Patients benefit from comprehensive evaluation, including detailed clinical examination, advanced imaging, electrophysiology, and genetic testing. This integrated approach allows for precise diagnosis and personalised care, ensuring that each patient receives management tailored to their specific condition and needs.
Importantly, these centres also play a role in connecting patients with research initiatives and clinical trials. Participation in such programmes can provide access to emerging therapies and contribute to the advancement of knowledge in this rapidly evolving field. For patients with inherited retinal disease, this offers a sense of hope that extends beyond current treatment options.
Despite these advances, challenges remain. The genetic complexity of IRD means that not all patients will have a clearly identifiable mutation, and not all identified mutations currently have targeted therapies. Furthermore, the variability in disease expression even among individuals with the same genetic mutation complicates prognosis and management. Continued research is essential to address these gaps and to translate scientific discoveries into clinical benefit.
Early diagnosis remains a key factor in improving outcomes. Patients presenting with symptoms such as night blindness, unexplained visual field loss, or a family history of retinal disease should be referred for specialist evaluation. Timely diagnosis allows for appropriate counselling, monitoring, and, where available, consideration of therapeutic interventions.
Education is equally important. Patients should be informed about the nature of their condition, expected progression, and available support services. Empowering patients with knowledge enables them to make informed decisions about their care and to adapt to the challenges posed by their condition.
From a broader perspective, inherited retinal disease highlights the intersection between genetics and clinical ophthalmology. It underscores the importance of a holistic approach that combines clinical expertise, advanced diagnostics, and compassionate patient care. As our understanding of the genetic basis of these conditions continues to grow, so too will our ability to diagnose, manage, and ultimately treat them.
In conclusion, inherited retinal disease represents a complex and heterogeneous group of conditions that are a major cause of visual impairment. Characterised by retinal dystrophy, often presenting as night blindness and evolving into progressive vision loss, these disorders require a thoughtful and multidisciplinary approach. Recognised as a form of family eye disease, they carry implications not only for the individual but also for their relatives.
With advances in genetic testing, imaging, and emerging therapies, the outlook for patients with inherited retinal disease is improving. Centres such as Barraquer Eye Hospital are at the forefront of this progress, offering comprehensive care and access to cutting-edge developments. While challenges remain, the integration of genetics into clinical practice is reshaping the future of retinal care, bringing new possibilities for diagnosis, management, and ultimately, treatment.
Email: appointments@barraquer.ae
Phone (outside UAE): +971 4 573 9999
Toll-Free (inside UAE): 800 234 823 (BEHUAE)
Working Hours & Location: Click on Google Map Link